2025 年医药健康领域法规迎来数字化升级关键期:《医疗器械生产质量管理规范》修订草案明确电子记录管理要求,NMPA《药品记录与数据管理要求》持续深化执行,FDA 21 CFR Part 11 合规标准仍是国际准入核心门槛。当 "真实、准确、完整、及时、可追溯" 成为电子记录的硬性指标,企业该如何快速响应法规变化,实现从纸质记录到数字化合规的高效转型?GMPEBR电子批记录系统给出了答案。
法规新风向:电子记录不再是 "选择题",而是 "必修课"
近期密集出台的行业法规,正在重塑医药、医疗器械领域的记录管理逻辑:
-
2025 版《医疗器械生产质量管理规范》首次系统性提出电子记录要求,章节从 "文件管理" 升级为 "文件和数据管理",明确权限控制、变更追溯等核心要求,2026 年 11 月 1 日起正式施行。
-
NMPA 明确电子记录与纸质记录具有同等法律效力,要求计算机(化)系统实现操作追溯、权限分离、数据备份等全生命周期管理。
-
国际层面 FDA 21 CFR Part 11 将数据完整性作为核心考核点,从系统安全到操作追溯形成全链条要求。
这意味着传统纸质记录 + 人工复核的模式,已难以满足法规对过程管控、数据追溯的精细化要求,数字化转型成为企业规避合规风险的必然选择。
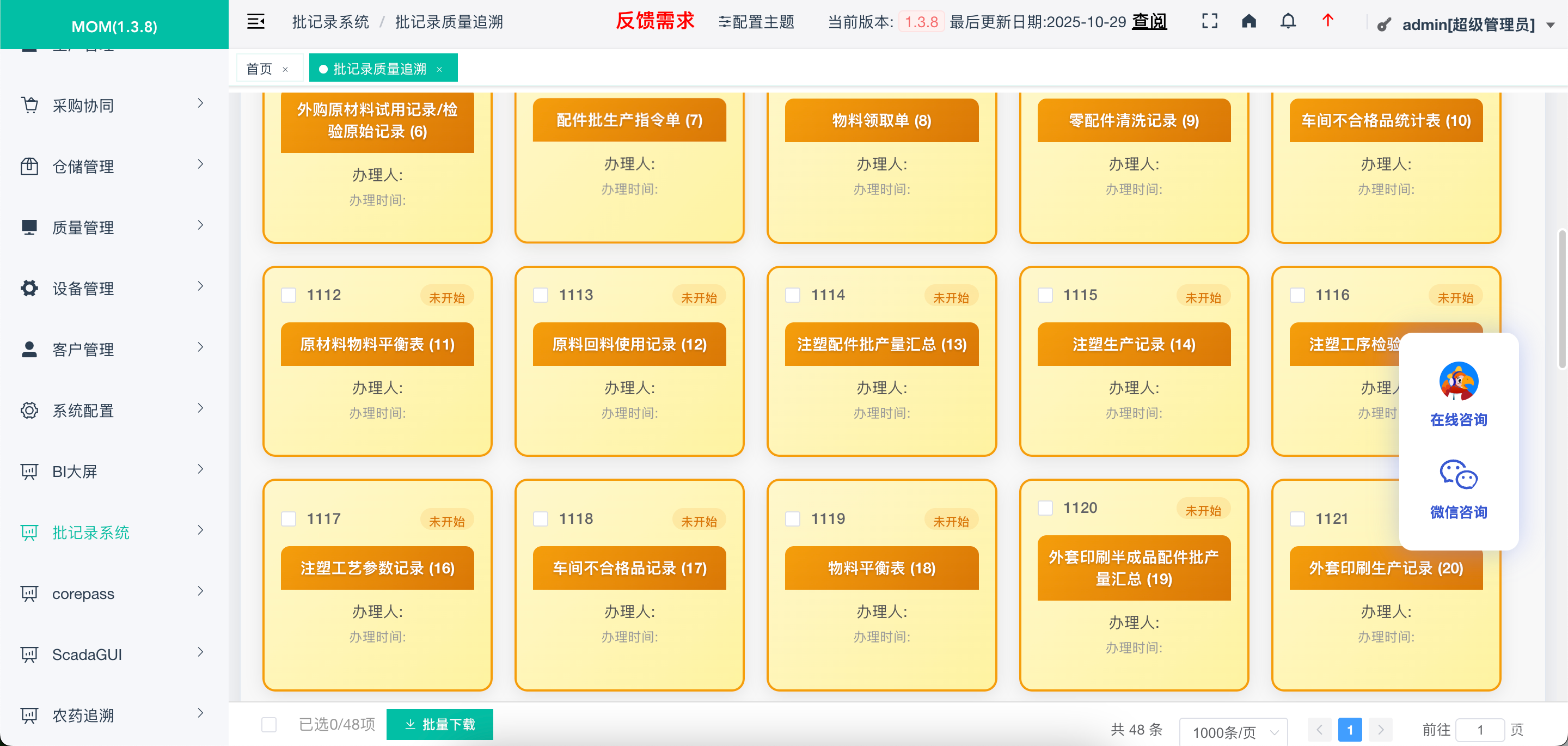
GMPEBR 一键电子批记录系统:精准匹配新规要求,让合规 "不踩雷"
作为贴合国内国际法规要求的电子批记录系统,GMPEBR 一键电子批记录系统 从功能设计到技术实现,全面对标新规核心要求,让合规管理更高效、更可靠:
全维度满足 "五性" 要求
严格遵循法规对电子记录 "真实、准确、完整、及时、可追溯" 的五性要求,系统自动采集生产过程数据,避免人工录入误差,同时通过时间戳精准记录每一步操作,确保数据全程可追溯,完全符合 2025 版医疗器械 GMP 及药品数据管理规范。
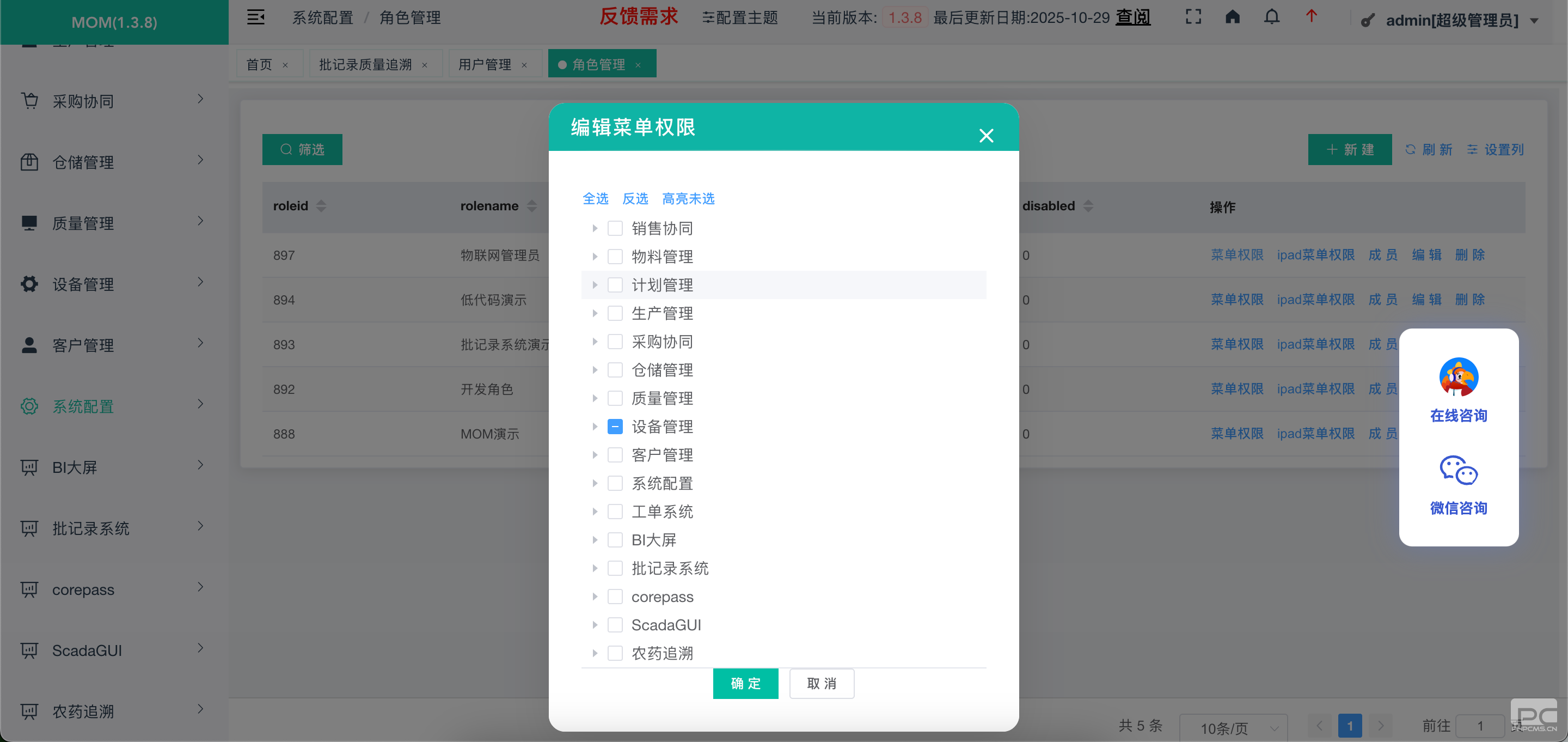
权限与签名合规设计
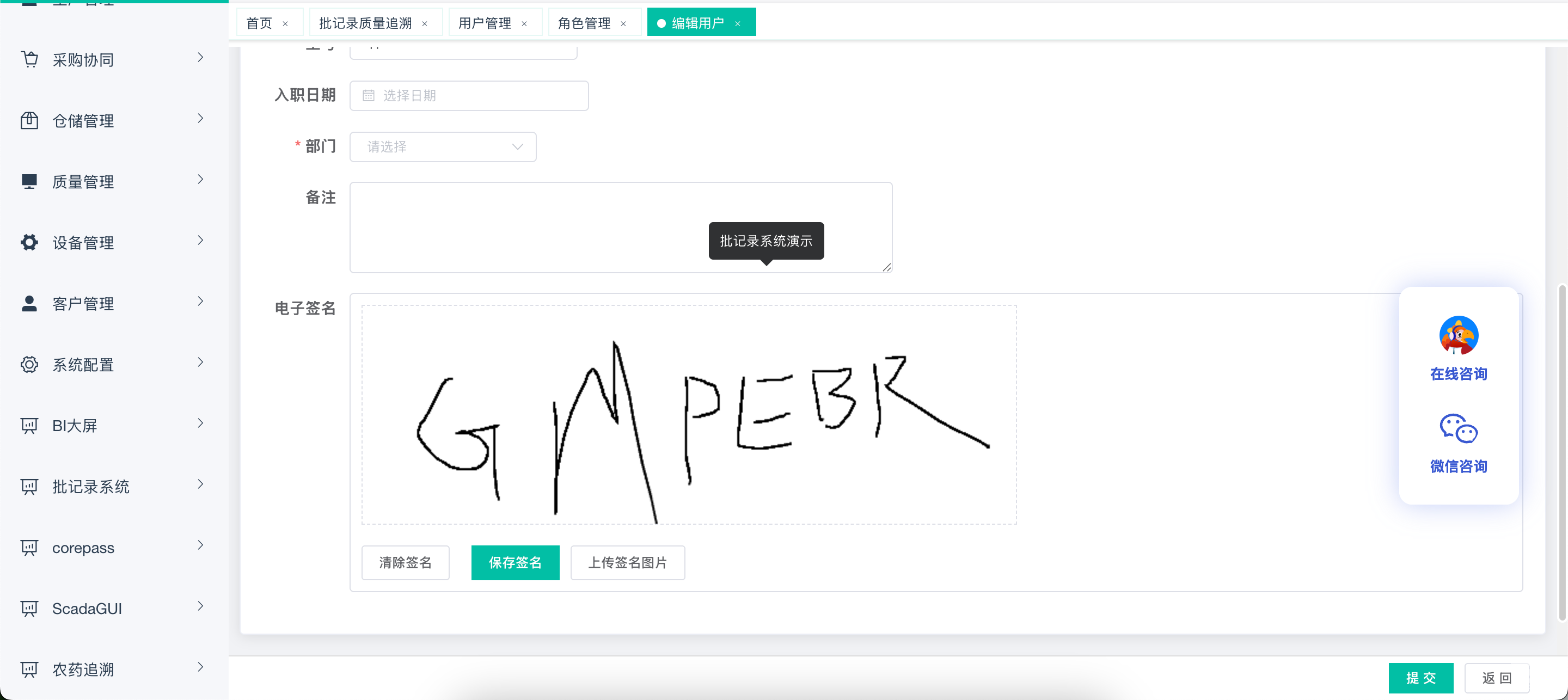
建立分级权限管理体系,实现操作与系统管理权限分离,业务负责人权限与职责精准匹配,杜绝越权操作。电子签名功能完全符合《中华人民共和国电子签名法》要求,确保签署人专有可控、改动可发现,满足 NMPA 与 FDA 双重合规标准。
全生命周期数据管控
覆盖电子记录生成、修改、归档全流程,任何数据变更、删除操作均自动记录审计轨迹,形成不可篡改的操作日志。系统支持数据定期备份与验证恢复流程,确保数据在规定保存期限内可查阅、可追溯,彻底解决 Excel 等传统格式易篡改、难追溯的合规痛点。
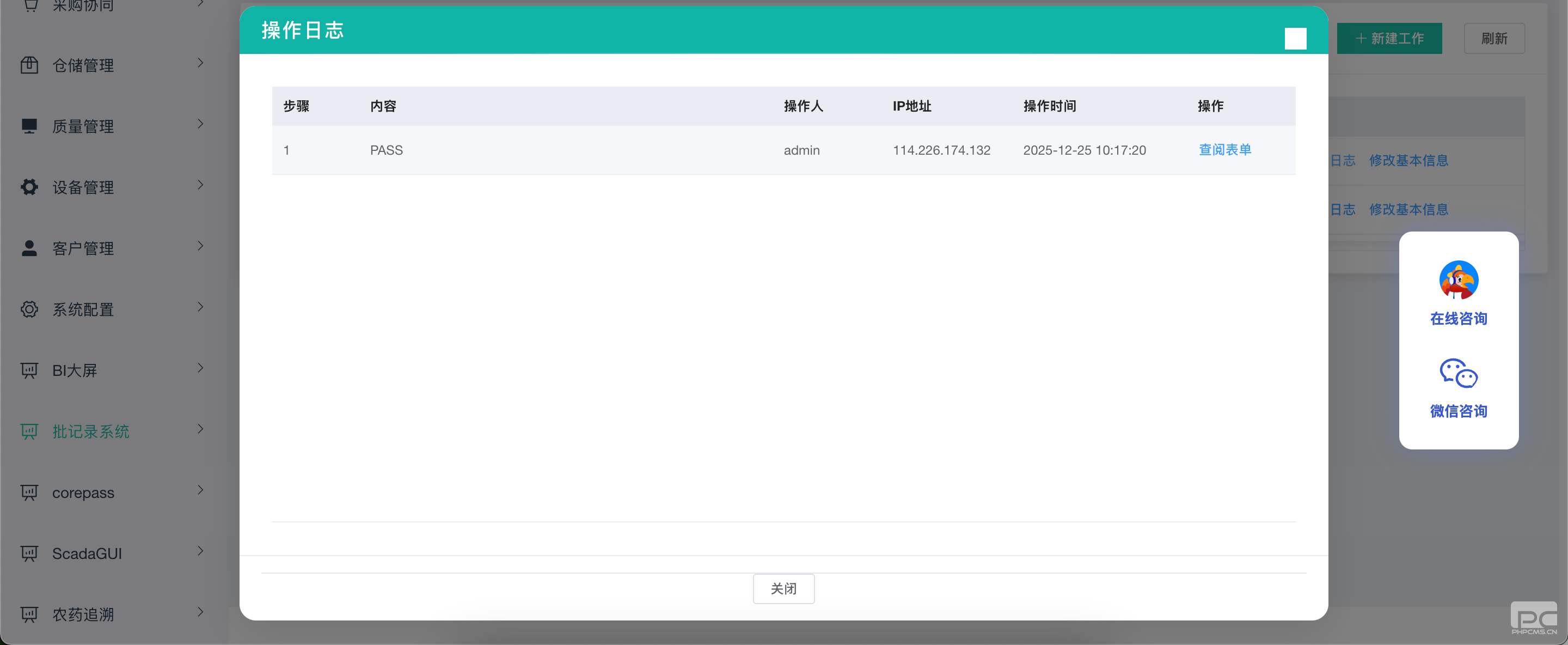
无缝衔接业务场景
针对医药、医疗器械生产流程特点,系统支持生产计划下达、过程执行记录、质量检验、成品放行等全流程数字化管理,数据实时流转、自动关联,减少人工传递误差。同时支持报告导出、打印功能,满足纸质备份与电子记录并存的过渡需求,实现合规与效率的双重提升。
不止于合规:以数字化驱动生产提质
GMPEBR 一键电子批记录系统的价值远不止满足法规要求,更能通过数字化手段为企业创造实际效益:
-
效率提升:替代人工手写记录与复核,数据录入效率提升 50% 以上,检索查询速度较纸质记录提升数十倍。
-
成本优化:减少纸质记录的打印、存储、管理成本,降低因记录失误导致的返工、合规处罚风险。
-
决策支撑:实时采集生产过程数据,形成可视化报表,为生产调度、质量改进提供数据支撑,助力精益生产。
结语:合规转型窗口期,选对系统少走弯路
2025-2026 年是医药健康行业数字化合规的关键过渡期,企业需在法规施行前完成系统升级与流程适配。GMPEBR 一键电子批记录系统以法规为纲、以业务为核,既解决当下合规痛点,又为未来数字化升级预留空间,是企业应对法规变化、提升核心竞争力的理想选择。