最近小编正在整理公司的一些批生产记录,为了理清楚相关细节和内容查阅了相关材料和标准。今天在这里和大家进行简单的分享,希望对大家在药品生产过程中遇到的批记录撰写问题有所助益。
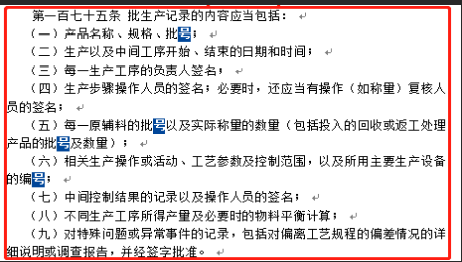
药品生产批生产记录是确保药品质量可追溯性的重要文件,其内容需严格遵循《药品生产质量管理规范》(Good Manufacturing Practices, GMP)及相关法规要求。那么我们就先看一下GMP中对批生产记录的相关描述,在第一百七十五条中详细规定了批生产记录所应包含的具体内容,分别是:产品名称、规格、批号;生产以及中间工序开始、结束的日期和时间;每一生产工序的负责人签名;生产步骤操作人员的签名;必要时,还应当有操作(如称量)复核人员的签名;每一原辅料的批号以及实际称量的数量;相关生产操作或活动、工艺参数及控制范围,以及所用主要生产设备的编号;中间控制结果的记录以及操作人员的签名;不同生产工序所得产量及必要时的物料平衡;对特殊问题或异常事件记录(包括对偏差情况的详细说明或调查报告)并经签字批准。
我国国家药品监督管理局已在2017年加入ICH(国际人用户药品注册技术协调会),这意味着我们国家药品生产向着国际化发展迈进了一大步。在ICH-Q7-6.5中指出:批生产记录是由批工艺规程产生出来的正确的批记录。同时对批生产记录的内容提了要求:日期,时间(必要时);主要设备的标识;每批的标识,包括原辅料、中间品或任何用于生产的再加工材料的批号、重量和容器具;关键工艺参数的实际值;任何取样操作;每个关键步骤的操作者和复核人的签名;中间控制的检查结果;适当阶段或时间的实际产率;中间体或API的包装和标签说明;API或中间体的商业标签(如可市售);发现任何偏差,偏差的评估、调查(必要时),以及单独存放的调查报告的参考;放行检查结果。法规全文可在CDE官网(ICH工作办公室下载)。ttps://www.cde.org.cn/ichWeb/guideIch/toGuideIch/1/0

那么批生产记录到底该怎么撰写呢?以下为我的个人理解,仅代表个人观点,仅作学习交流用。
1.批生产指令单
按照批生产计划下达批生产指令,按照批生产指令发放批生产记录。
批生产指令应包含以下信息:要生产的产品的名称、代码、批号、批量、关键步骤可接受收率范围、文件编号及版本号、中间控制可接受标准等;设备一览表;备料指令和记录(每种物料或耗材的名称、物料代码、厂家批号、本厂批号、质量标准、处方量、备料量);批生产操作要求;签发人、日期;审核人、日期;批准人、日期。
2. 生产过程记录
(1)产品名称、产品代码、批号、规格(若适合)、生产起止日期。
(2)员工签名对照表:姓名、签名、日期。
(3)关键工序收率标准及汇总表(在新产品刚开始生产时,可将其范围定宽一些以防出现偏差,随后工艺稳定、经验累积可逐步提升收率)和批产范围。
(4)涉及生产过程的记录:最好按照每个步骤一部分来设计,每个步骤再根据实际情况划分不同单元过程。
以生物药纯化批生产记录为例,比如可以分为生产前检查(包括公共系统、厂房、设施设备、物料、耗材等几个部分的检查)、容器具准备(清洁、干烤、湿热灭菌等根据实际情况设置)、分离纯化(亲和层析、低pH病毒灭活、阴离子层析、阳离子层析、除病毒过滤、超滤、除菌过滤等)、附录(生产过程中涉及到的需要粘贴的标签或记录等)。
其中每个单元过程又需要包含指令(参数描述或者操作描述)以及记录(应与指令一一对应)、操作人员及复核人员(如需要)签名和日期以及起止时间。操作描述最好不要用约、接近等词汇,设定好上下范围;关键步骤(或单元过程)如需要中间控制要有可接受标准并记录实际值;操作范围不宜太窄,会使可操作性降低;关键工艺参数要记录实际值以备追溯;取样要有记录(样品名称、取样量、取样时间、取样人、用途等);涉及计算的需要有计算公式。
每个步骤或每个单元过程(依据重要程度决定)结束后,应该有一个可以记录检查步骤完成情况的记录,是否有偏差,如有偏差应记录其调查、报告以及附单独的偏差记录等,并由工序主管及QA签字确认。
(5)每个房间生产结束后都要有清场记录,包括有清场日期、时间、有效期、清场人及复核人签名和日期,经工序主管及QA检查合格签字方可。